




Meta把内部设计系统开源了:支撑内部13000+应用:专为Agent调优
2026-07-03
2026-07-09 0


在有机合成和药物化学中,羧酸、醇和胺是最常见的三类官能团。相比传统使用的苄基溴、苄基金属试剂等偶联前体,苄胺来源丰富、稳定性更高、储存方便,尤其是杂环苄胺在药物分子中十分常见。然而,苄胺作为烷基自由基前体参与C(sp³)–C(sp³)键构建的研究却相对有限。
目前胺与羧酸最常见的反应仍然是形成酰胺键,而直接将二者转化为新的碳碳键的方法十分稀少。苄胺与醇之间的直接偶联更是鲜有报道。因此,开发一种能够利用天然存在的苄胺、羧酸和醇作为原料,实现高效C(sp³)–C(sp³)偶联的新策略,对于药物分子和复杂骨架的快速构建具有重要意义。
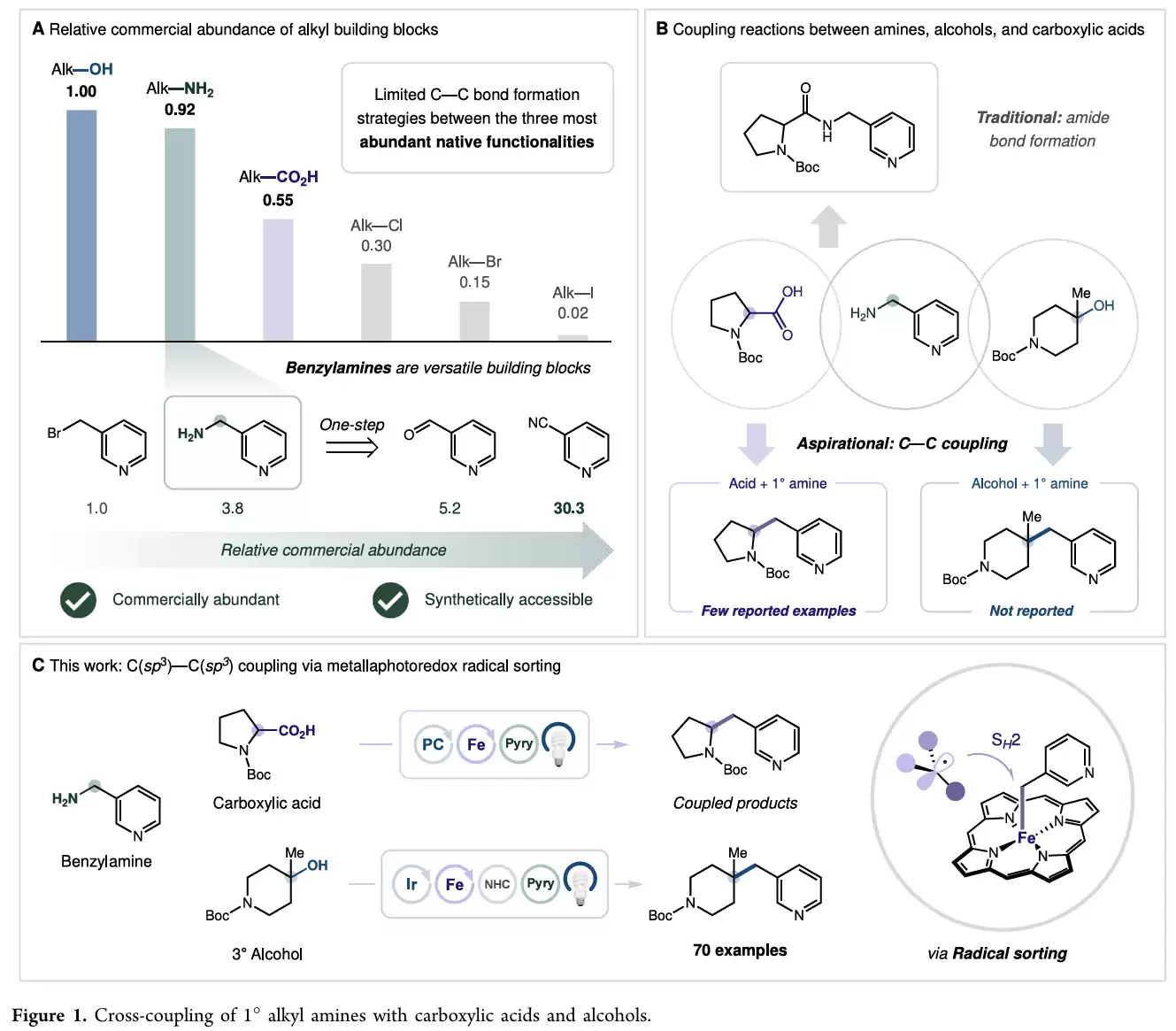
MacMillan团队提出了一种金属光氧化还原催化体系,将苄胺与羧酸或叔醇进行脱胺交叉偶联。反应设计的核心在于自由基分类策略。研究人员先将苄胺转化为Katritzky盐,在光催化条件下生成苄基自由基。同时,羧酸经过脱羧或叔醇经过脱氧过程生成三级烷基自由基。
如果体系中同时存在两种自由基,往往容易发生随机偶联而导致选择性下降。为解决这一问题,作者引入Fe-卟啉催化剂。该催化剂优先捕获苄基自由基形成Fe–C中间体,随后三级烷基自由基通过SH2过程进攻铁中心,最终实现定向交叉偶联。这种策略避免了传统镍催化中常见的氧化加成和转金属化步骤,也有效抑制了自由基自偶联副反应。
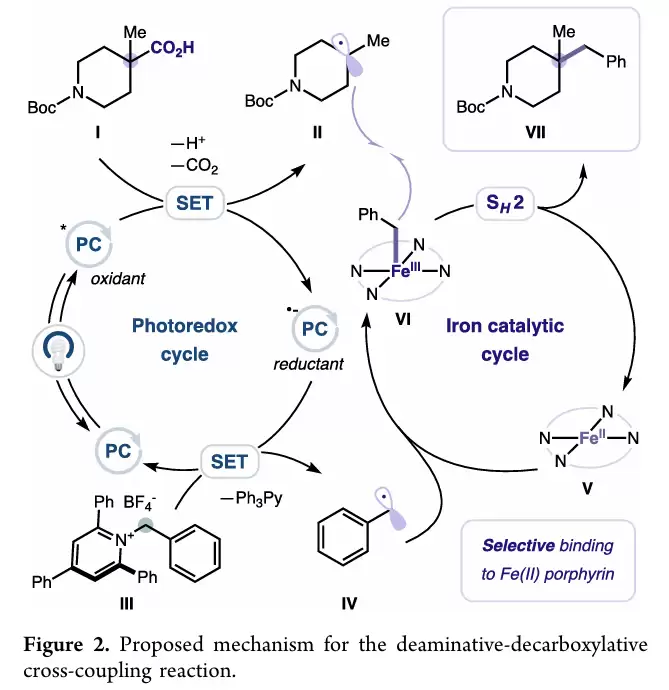
作者提出的机理可以概括为三个关键步骤。
羧酸在碱作用下形成羧酸盐,经激发态光催化剂氧化后迅速脱羧,得到三级烷基自由基。与此同时,Katritzky盐接受电子后发生β断裂,释放苄基自由基。
生成的苄基自由基首先被Fe(II)-卟啉捕获,形成Fe(III)-苄基物种。随后三级烷基自由基与该铁中间体发生双分子均裂取代反应,从而形成新的C(sp³)–C(sp³)键,并再生铁催化剂。
控制实验中检测到了苄基自由基二聚副产物,这为苄基自由基中间体的存在提供了直接证据,也支持了作者提出的机理模型。
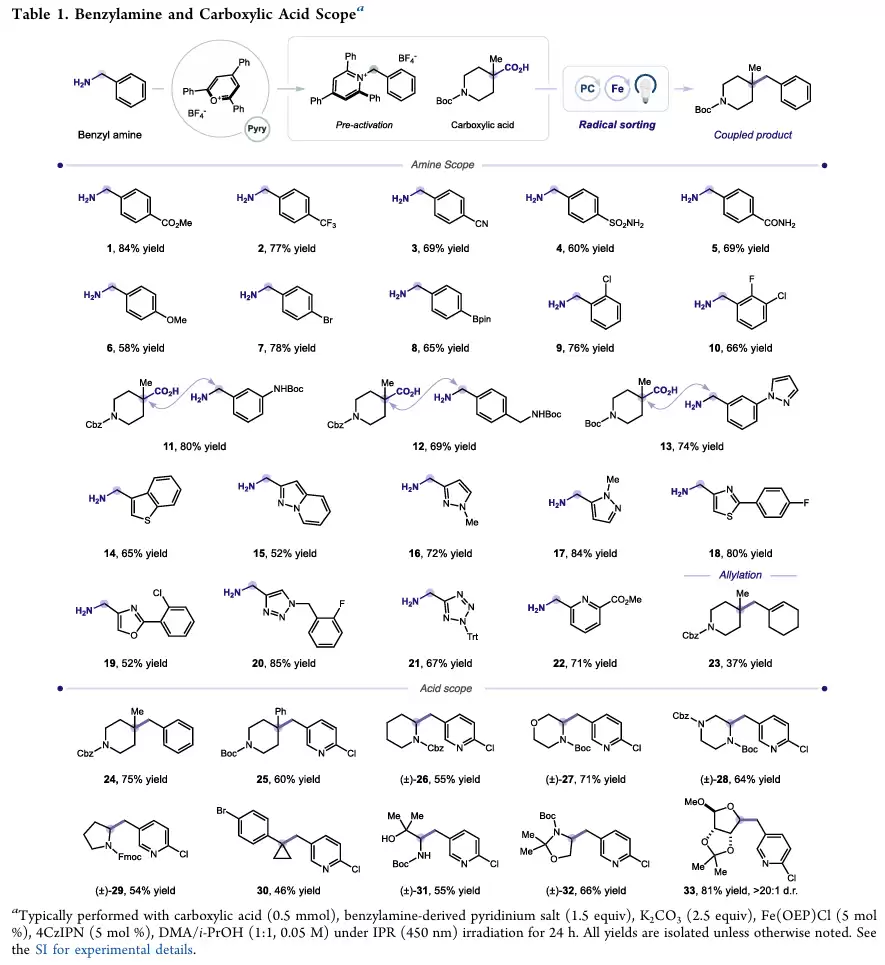
在苄胺底物考察中,无论是给电子还是吸电子取代基都能够获得较高收率。酯基、磺酰胺、溴原子、硼酸酯等官能团均能耐受反应条件。
多种药物化学中常见的五元杂环苄胺均表现出良好的兼容性,包括吡唑、咪唑、四唑等结构。四唑底物的成功转化尤其具有意义,因为四唑常被视为羧酸的生物电子等排体。
在羧酸部分,α-三级羧酸、氨基酸衍生物以及糖类衍生物均可顺利发生偶联。部分底物还能以优异的非对映选择性生成目标产物,显示出该方法在手性分子构建方面的潜力。
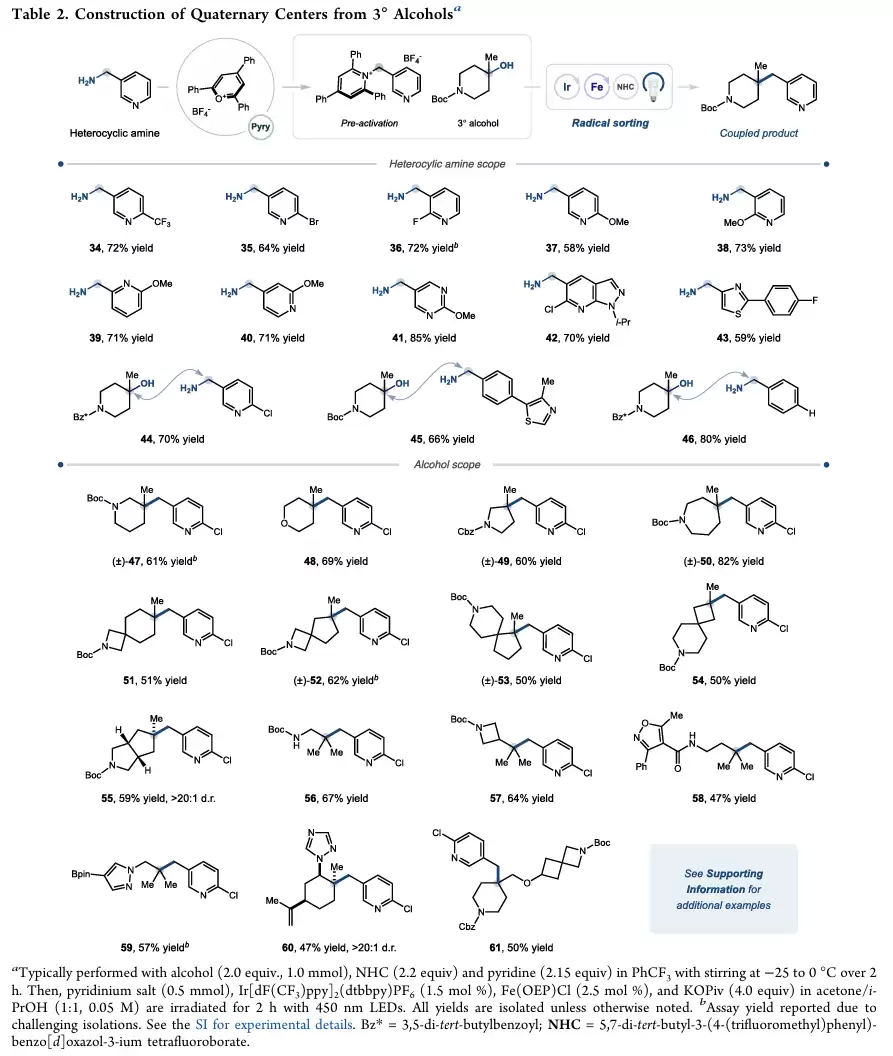
除了羧酸之外,作者进一步将反应扩展到叔醇体系。叔醇在苯并噁唑鎓活化剂作用下原位转化为自由基前体,随后经过光氧化生成三级烷基自由基。
研究发现,各种环状叔醇、螺环叔醇以及稠环叔醇均能获得良好收率。即使面对较大的空间位阻和较高的环张力,反应仍然能够顺利进行。通过该方法可以高效构建全碳季碳中心,而这类结构长期以来都是药物化学和天然产物合成中的重要挑战。相比之下,一、二级醇则难以适用于该体系,这与相应自由基亲核性较弱有关。
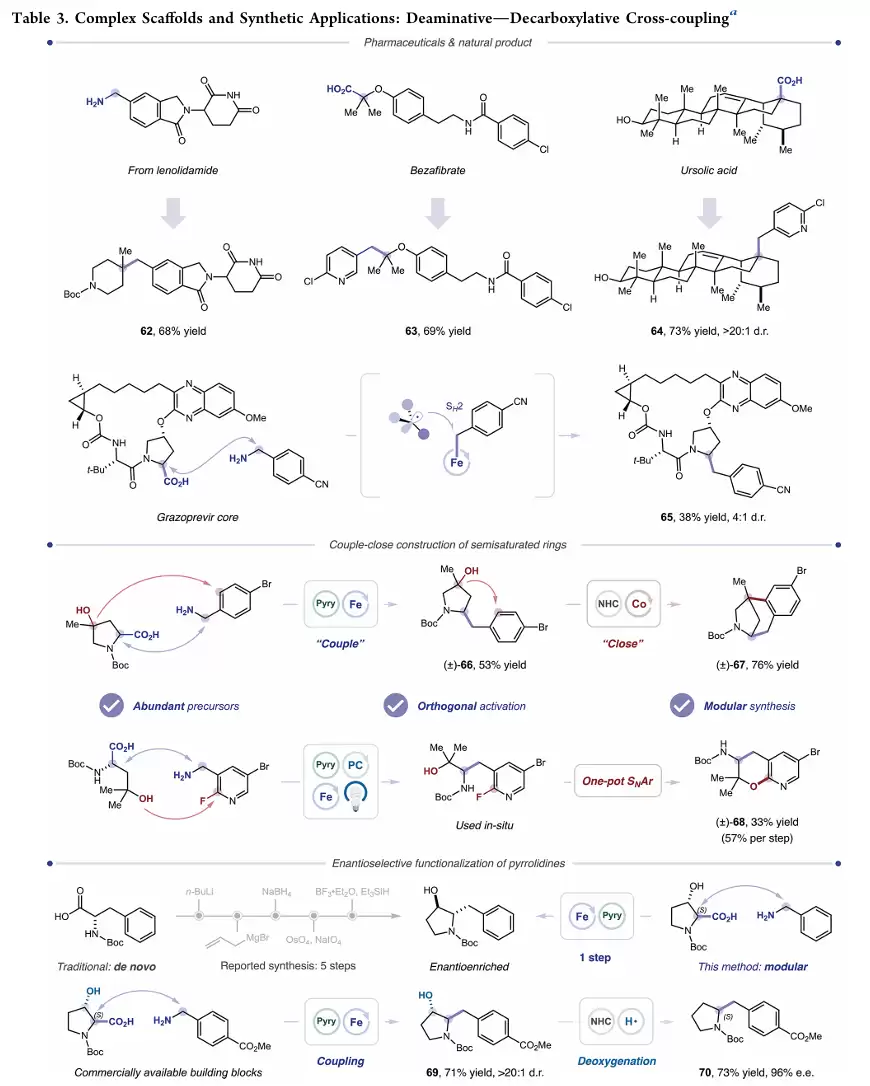
该工作首次实现了苄胺与叔醇之间的直接脱胺C(sp³)–C(sp³)交叉偶联,同时建立了苄胺与羧酸偶联的新方法。反应利用廉价易得的天然官能团作为偶联原料,通过光催化与铁卟啉催化协同作用,实现了SH2自由基分类控制下的高选择性碳碳键构建。方法学具有广泛的底物兼容性,能够耐受复杂杂环、药物分子以及糖类衍生物,并成功应用于复杂活性分子修饰、半饱和芳环骨架构建以及手性吡咯烷衍生物合成。
文章链接
https://doi.org/10.1021/jacs.6c04676
Copyright(C) 2020-2026 jiyx.com All Rights Reserved 联系方式:[email protected]